


Protegemos tu salud a través de la Ciencia
Desde el ISCIII mostramos nuestra solidaridad con todas las personas afectadas por el desastre de la DANA
Todo nuestro apoyo en estos momentos tan difíciles






En el

Hallado un nuevo gen causante de una enfermedad neurológica ultra-rara
El descubrimiento de un segundo gen implicado en el desarrollo del síndrome de Kleefstra, enfermedad rara que afecta al desarrollo del cerebro, abre las puertas a avances en el diagnóstico. Saber más
Tecnologías de vanguardia para identificar enfermedades raras sin diagnóstico
El Programa SpainUDP del IIER-ISCIII impulsa la identificación de enfermedades raras complejas. Este miércoles es el Día Mundial de las Personas sin Diagnóstico. Saber más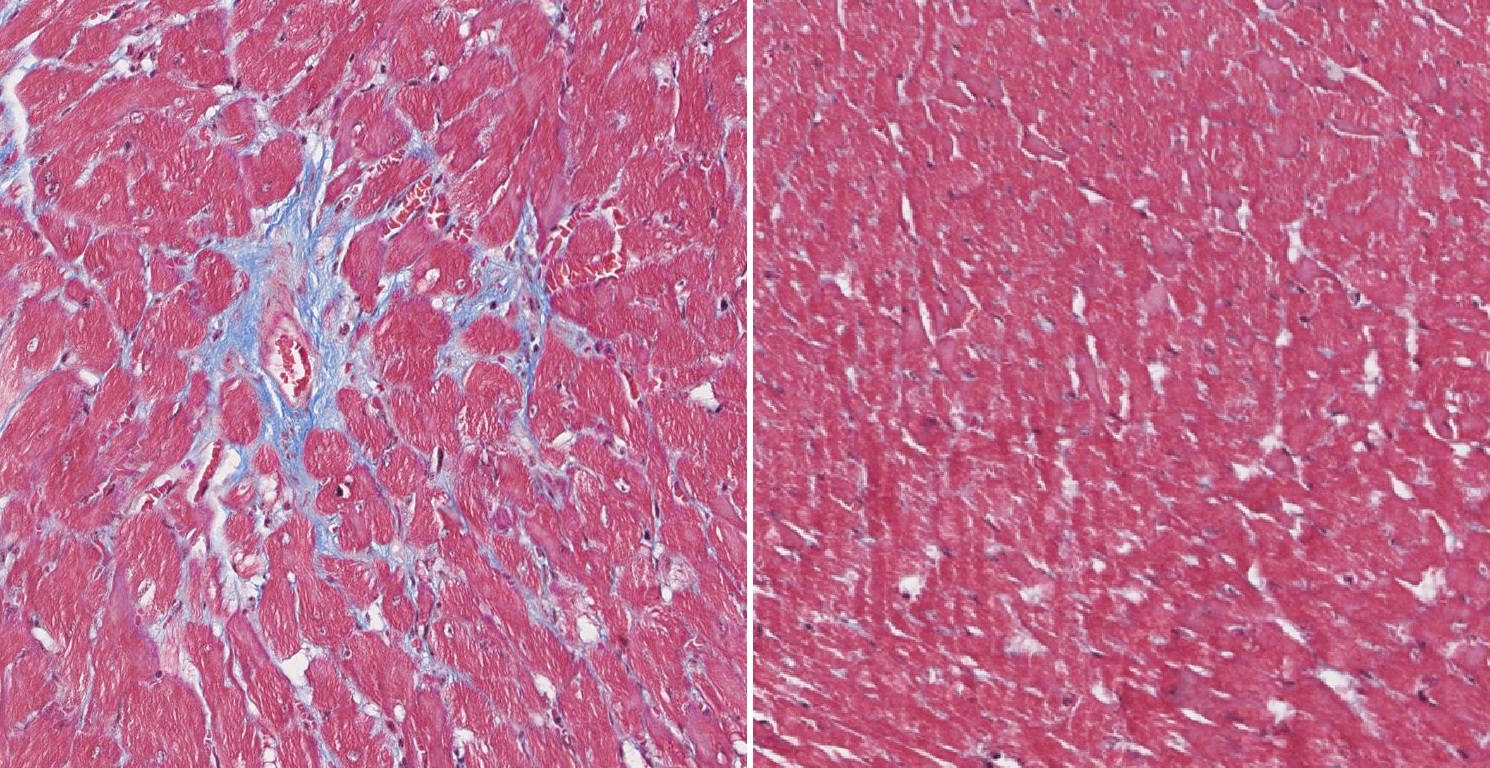
Primeros avances hacia una terapia génica con CRISPR para una enfermedad rara muscular sin cura
La técnica de edición genética CRISPR ofrece resultados prometedores para la distrofia muscular congénita asociada al gen LMNA. Hoy se celebra el Día Mundial de las Enfermedades Raras Saber más1/3
Actualidad
Identificadas alteraciones en un nuevo gen causante de una enfermedad neurológica ultra-rara
Una investigación internacional co-liderada desde el Instituto de Salud Carlos III (ISCIII) ha identificado alteraciones genéticas, hasta ahora desconocidas, relacionadas con la aparición del síndrome de Kleefstra, una enfermedad rara que afecta al desarrollo neurológico. Los resultados del estudio se han publicado en la revista Nature Communications. La doctora María José Barrero, del Instituto de Investigación de Enfermedades Raras (IIER) del ISCIII y líder de la participación española, explica que estos hallazgos pueden cambiar la forma de diagnosticar la enfermedad y abrir la puerta al estudio de terapias para tratar sus principales manifestaciones clínicas, relacionadas con dificultades cognitivas, del lenguaje y del comportamiento. Hace veinte años se demostró que el síndrome de Kleefstra está causado por alteraciones en el gen EHMT1. Ahora, la investigación coliderada desde el ISCIII ha descubierto que mutaciones en otro gen de función similar, denominado EHMT2, también causan enfermedad. Este gen, cuando está alterado y manifiesta variantes patógenas, modifica la función de G9a, una enzima implicada en la regulación epigenética de la expresión génica. Las alteraciones se relacionan con la aparición de un mecanismo dominante negativo que causa un nuevo síndrome del neurodesarrollo con características similares al síndrome de Kleefstra. Hasta este momento este gen no se había asociado con el desarrollo de ninguna enfermedad, lo que supone un gran avance para el diagnóstico de este nuevo síndrome asociado a alteraciones en EHMT2. La investigación ha contado con financiación del Ministerio de Ciencia, Innovación y Universidades, a través de convocatorias del ISCIII y de la Agencia Estatal de Investigación, con apoyo de fondos europeos, de la Fundación Inocente y de la asociación de pacientes Kleefstra España. Colaboración internacional y Programa de Casos sin Diagnostico La doctora Barrero explica que este estudio internacional "es fruto de un trabajo colaborativo con la Charles University en Praga que ha reclutado pacientes en varios países europeos, entre ellos España, y en Estados Unidos, y en el que han participado numerosos hospitales y centros de investigación nacionales e internacionales". En concreto, han colaborado equipos de 29 entidades científicas de 7 países, con participación de dos hospitales españoles. El estudio se enmarca en la labor del Programa de Casos sin Diagnostico SpainUDP, que coordina el ISCIII, que permitió diagnosticar el primer caso en España de la enfermedad. También han participado en el estudio otras dos plataformas coordinadas desde el ISCIII: el Registro de Pacientes de Enfermedades Raras (RePER) y el Biobanco Nacional de Enfermedades Raras (BioNER). Foto de grupo en uno de los laboratorios del IIER-ISCIII. En la primera fila: Beatriz Martínez Delgado, Maria Jose Barrero y Eva Bermejo. Arriba: Sheila Ramos, Diana Sánchez Ponce, Beatriz Baladrón, Esther Hernández, Marta Fernández, Javier Alonso y Gema Gómez. En concreto, análisis in vitro y en modelos animales han revelado que las alteraciones producen retraso del crecimiento, dismorfia facial y craneal, y comportamientos anómalos, síntomas que forman parte del cuadro clínico que se observa en las personas afectadas. La única familia española afectada, y participante en el estudio, señala que esta investigación supone un punto de inflexión: "Nos permitió llegar al diagnóstico de nuestro hijo y comprender por fin su situación. Para nosotros, la investigación significa todo, abre una verdadera esperanza hacia futuros tratamientos. La colaboración entre pacientes, familias, asociaciones y comunidad investigadora es esencial para avanzar". La investigadora por ISCIII explica que la patogénesis -el origen de la enfermedad- de la disfunción genética de EHMT2 que causa la patología humana “sigue siendo en gran medida desconocida, por lo que la identificación en pacientes con Kleefstra de siete variantes alteradas en el gen EHMT2 supone un paso importante para entender los mecanismos de la enfermedad y seguir mejorando el diagnóstico". Con este fin, la Unidad de Modelos y Mecanismos del IIER continúa investigando sobre este síndrome con el apoyo de la federación Española de Enfermedades Raras (FEDER). • Referencia del artículo: Hnízda, A., Martinez-Delgado, B., Sanchez-Ponce, D. et al. De novo EHMT2 variants cause an autosomal dominant EHMT2-related Kleefstra syndrome via loss of G9a methyltransferase activity. Nat Commun (2026). https://doi.org/10.1038/s41467-026-74987-w. Noticias relacionadas: - Proyecto pionero para estudiar el ‘mapa del cerebro’ en el síndrome de Kleefstra
Saber másTecnologías de vanguardia para impulsar la identificación de enfermedades raras sin diagnóstico
El Instituto de Salud Carlos III (ISCIII), a través de su Instituto de Investigación de Enfermedades Raras (IIER), lleva años desarrollando un programa para mejorar el diagnóstico de patologías poco comunes especialmente difíciles de identificar. El Programa SpainUDP, creado en 2013, coordina la investigación y análisis de casos clínicos complejos para alcanzar un diagnóstico que es fundamental para superar barreras clínicas, sanitarias y sociales. El Día Mundial de las Personas Sin Diagnóstico, que se conmemora cada 29 de abril y que complementa el Día Mundial de las Enfermedades Raras (28 de febrero), es una oportunidad para visibilizar la necesidad de seguir impulsando la investigación para mejorar la identificación de patologías poco comunes difíciles de catalogar. Según explica Eva Bermejo, directora del IIER-ISCIII, "muchas personas afectadas, y sus familiares, esperan en ocasiones años e incluso décadas, hasta que la ciencia puede identificar la enfermedad que explique su condición". Según añade, la ausencia de diagnóstico dificulta el acceso a tratamientos adecuados, empeora la calidad de vida y supone una importante barrera sociosanitaria. Desde su nacimiento hace 13 años, el Programa SpainUDP -coordinado por las investigadoras Beatriz Martínez y Estrella López, junto con Eva Bermejo- ha recibido 647 propuestas de casos clínicos complejos para, en coordinación con los especialistas clínicos, tratar de acelerar un diagnóstico y facilitar la calidad de vida de las personas afectadas. Más de la mitad de las propuestas recibidas (373, casi un 58% del total) han sido aceptadas al cumplir los requisitos para su estudio y, de este total, más del 60% (227 casos) han sido ya completamente evaluadas (el resto están pendientes de nuevas pruebas o información adicional). De los casos analizados, que incluyen casos pediátricos y adultos, casi la mitad (108) han logrado finalmente un diagnóstico genético. SpainUDP ha diagnosticado el 52,5% de los casos pediátricos. Estos resultados han sido posibles gracias a la amplia red de colaboraciones nacionales e internacionales. Entre las colaboraciones nacionales destaca la red de clínicos directamente implicados en la atención a estos pacientes, ya que actúan como apoyo esencial para SpainUDP desde el tejido asistencial del Sistema Nacional de Salud. En el caso internacional, el partenariado europeo ERDERA ha comenzado este año un proceso de reanálisis sistemático de casos sin diagnóstico por toda Europa, que incluye pacientes procedentes del Programa SpainUDP. Esta labor de estudio permite reevaluar datos genéticos a la luz de nuevos conocimientos y herramientas tecnológicas, lo que permite aumentar las posibilidades diagnósticas. En este sentido, el IIER-ISCIII ha incorporado en los últimos años el uso de tecnologías de vanguardia para el análisis clínico y genético. Entre las incorporaciones más recientes destacan el mapeo óptico del genoma (OGM), la secuenciación del genoma mediante lecturas largas (long-read sequencing) y el estudio específico de genes RNU, recientemente identificados como una de las causas más frecuentes de síndromes del neurodesarrollo. Estos avances han permitido alcanzar diversos diagnósticos en casos no resueltos, entre ellos el diagnóstico del primer caso recibido en SpainUDP, en 2013. De izquierda a derecha: María José Barrero, Victoria Moneo, Esther Hernández, Marta Fernández, Sheila Ramos, Sergio Casas, Eva Bermejo, Estrella López, Beatriz Martínez, Raúl Contreras, Gema Gómez y Beatriz Baladrón, del IIER e implicados en el Progrma SpainUDP. En la imagen no aparece Lidia Mirela, que también forma parte del proyecto. En esta labor de impulso diagnóstico, el Programa SpainUDP integra diferentes estrategias para evaluar, mediante pruebas funcionales, el papel de nuevos genes que puedan ser causantes de enfermedad, pero que son actualmente desconocidos. Para ello, en la unidad de Modelos y Mecanismos liderada por Maria José Barrero, que también se integra en SpainUDP, se llevan a cabo iniciativas como el desarrollo en laboratorio de nuevos modelos celulares in vitro y la generación de modelos de organoides, mini-órganos artificiales que imitan el funcionamiento de órganos reales, sobre los que se pueden realizar las pruebas más adecuadas para cada caso. También en la Unidad de Modelos de Enfermedad en Drosophila, liderada por Sergio Casas, se estudian variantes genómicas y su funcionalidad en avatares creados en dicho organismo. De esta manera se mejora el estudio de la alteración de genes específicos como causantes de nuevos síndromes, por ejemplo en enfermedades del neurodesarrollo. Las coordinadoras del Programa SpainUDP en el IIER-ISCIII explican que este tipo de avances "pone de manifiesto que, incluso tras años de incertidumbre, la investigación y la innovación científica, y la revisión permanente con nuevas técnicas, de los casos que siguen sin identificar, pueden abrir nuevas vías para lograr un diagnóstico final. Para muchas personas la respuesta científica puede tardar, pero la investigación permite nuevos conocimientos que acercan el final de esa odisea por el desierto que, para algunas personas, puede suponer la búsqueda de diagnóstico para su enfermedad".
Saber más
Primeros avances hacia una terapia génica con CRISPR para una enfermedad rara muscular sin cura
Una investigación del Instituto de Salus Carlos III (ISCIII) ha utilizado la herramienta de edición genética CRISPR para explorar un posible tratamiento para una enfermedad muscular muy poco común y sin cura, la distrofia muscular congénita asociada al gen LMNA (abreviada como L-CMD). El estudio, liderado por un equipo del Instituto de Investigación de Enfermedades Raras (IIER), se ha publicado en la revista Molecular Therapy Advances. Los resultados de este trabajo, llevado a cabo en modelos celulares y animales, sugieren un aumento de la supervivencia en esta patología sin cura, y representan la primera validación terapéutica de una estrategia de edición génica mediada por CRISPR-Cas9 para tratar la L-CMD. La distrofia muscular congénita asociada al gen LMNA es un trastorno genético raro, actualmente incurable, que se caracteriza por debilidad muscular de aparición temprana, problemas cardiacos como la miocardiopatía dilatada e insuficiencia respiratoria. Se trata de una patología monogénica, es decir, causada por mutaciones en un solo gen. La edición genética CRISPR, especialmente prometedora para tratar este tipo de patologías, se basa en un mecanismo de ‘tijeras moleculares’ para modificar el ADN con precisión; permite cortar, eliminar o reemplazar secuencias específicas del genoma, lo que abre grandes vías para tratar enfermedades genéticas. El equipo del ISCIII, liderado por el doctor Ignacio Pérez de Castro, ha evaluado el potencial de CRISPR para eliminar la mutación más frecuentemente asociada a esta enfermedad, Lmna c.745C>T, p.R249W, utilizando para ello una guía específica, denominada sg745T. En modelos celulares, portadores de la citada alteración genética, han confirmado que el complejo Cas9/sg745T muestra una actividad específica hacia este alelo mutado dependiente de la dosis utilizada. Tras los resultados in vitro, análisis en un modelo de ratón confirmaron el potencial de esta estrategia gracias a la administración de la terapia usando virus adenoasociados en ratones portadores de la mutación causante de la enfermedad. La terapia aplicada a estos avatares murinos de pacientes humanos mejoró de manera significativa la patología cardiaca asociada a la enfermedad, lo que permitió incrementar la supervivencia media de los animales en más de un 20%. Este resultado adquiere especial relevancia si se considera que menos del 10% de los cardiomiocitos fueron editados por la maquinaria CRISPR en los animales tratados. De izda a dcha: Mercedes Dessy (Investigadora postdoctoral), Mónica Ferreiro (estudiante de grado, TFG), Mario Santafé (estudiante de doctorado), Iván Hernández (técnico especializado), Maria Luisa Martínez (técnica superior especializada), Ignacio Pérez de Castro (Jefe del grupo de Terapia Génica del IIER-ISCIII, investigador científico), Déborah Gómez (investigadora postdoctoral), Paula Esteban (estudiante de doctorado) y Alfredo Corral (estudiante de grado, TFG). El doctor Pérez de Castro explica que este estudio supone un primer paso hacia el desarrollo de una terapia capaz de aumentar la supervivencia y, potencialmente, curar la L-CMD. Asimismo, refuerza el potencial de la terapia génica basada en CRISPR para el abordaje de enfermedades raras como las laminopatías (enlace a esta noticia previa). "Aunque la eficiencia de edición obtenida hasta ahora es limitada, los resultados son prometedores y han superado nuestras expectativas. Ya estamos trabajando para superar estas barreras mediante el uso de editores evolucionados y la optimización de los sistemas de entrega de los complejos CRISPR. Nuestro objetivo es desarrollar una terapia de precisión que beneficie al mayor número posible de pacientes con esta enfermedad rara y que, además, sirva como plataforma para futuros tratamientos personalizados dirigidos a portadores de otras mutaciones en el gen LMNA o a pacientes con distrofias musculares causadas por mutaciones puntuales en otros genes". • Referencia del artículo: CRISPR-mediated targeting of the LMNA c.745C>T mutation enhances survival and cardiac function in congenital muscular dystrophy. Gómez-Domínguez, Déborah et al. Molecular Therapy Advances, Volume 34, Issue 1, 201653. https://www.cell.com/molecular-therapy-family/advances/fulltext/S3117-387X(25)00003-5. Vídeo divulgativo sobre la investigación y sus principales resultados. Youtube ISCIII.
Saber másAnota estas fechas
17 oct
17 sep
Empleo
IERPY 122-25-1-2-M3 (IND)
Inicio de plazo: 28/04/2026
Fin de plazo: 12/05/2026
Clase de personal: Laboral
Procedimiento / Modalidad: Indefinido (Art. 23 bis LCTI)
IERPY 122/25-BIS-M3-1 INDEFINIDO; INTPY 362/22- M3-4 y 5 INDEFINIDO
Inicio de plazo: 03/12/2025
Fin de plazo: 17/12/2025
Clase de personal: Laboral
Procedimiento / Modalidad: Indefinido (Art. 23 bis LCTI)
COOPERA 3M3y2M2 (IND)
Inicio de plazo: 19/11/2025
Fin de plazo: 02/12/2025
Clase de personal: Laboral
Procedimiento / Modalidad: Indefinido (Art. 23 bis LCTI)